De novo мутации в гистон-модифицирующих генах как одна из общих причин генетических заболеваний. Мутация - это неверный код. Норма и диапазон реакции
Все белки тела, записана в клеточной ДНК. Всего лишь 4 вида нуклеиновых оснований - и бесчисленное количество сочетаний аминокислот. Природа позаботилась о том, чтобы каждый сбой не был критичным и сделала избыточным. Но иногда искажение все же закрадывается. Его называют мутация. Это нарушение в записи кода ДНК.
Полезные - редки
Большинство таких искажений (более 99%) несут негативное значение для организма, что делает теорию эволюции несостоятельной. Оставшийся один процент не в состоянии обеспечить преимущество, так как далеко не каждый мутировавший организм дает потомство. Ведь в природе право размножаться имеют далеко не все. Мутация клеток чаще происходит у самцов - а самцы, как известно, в природе чаще умирают, не дав потомства.
Виноваты женщины
Впрочем, человек - исключение. У нашего вида чаще всего запускается безответственным поведением самок. Курение, алкоголь, наркотики, ЗППП - и ограниченный запас яйцеклеток, подвергающихся негативному воздействию с раннего детства. Если для мужчин существует, то для женщин даже небольшой стакан может спровоцировать нарушения правильного формирования яйцеклеток. Пока европейские женщины наслаждаются свободой, арабки воздерживаются - и рожают здоровых детей.

Неверно записано
Мутация - это постоянное изменение в ДНК. Оно может затрагивать небольшую зону или целый блок в хромосоме. Но даже минимальное нарушение сдвигает код ДНК, заставляя синтезировать совсем другие аминокислоты - следовательно, весь белок, кодируемый данным участком, будет неработающим.
Три типа
Мутация - это нарушение одного из видов - либо унаследованное, либо мутация de novo, либо локальная мутация. В первом случае это Во втором - нарушение на уровне сперматозоида или яйцеклетки, а также последствие воздействия опасных факторов после оплодотворения. Опасные факторы - это не только вредные привычки, но и неблагоприятная экологически обстановка (в том числе радиационная). De novo мутация - это нарушения во всех клетках тела, так как она возникает из ненормальных исходных. В третьем случае локальная, или возникает не на ранних этапах и затрагивает не все клетки тела, с большой долей вероятности она не передается потомству, в отличие от первого и второго типа нарушений.

Если проблемы возникли на ранних стадиях беременности, то возникает мозаичное нарушение. В этом случае часть клеток оказывается затронутыми болезнью, часть - нет. При этом виде высока вероятность, что ребенок родится живым. Большинство же генетических нарушений увидеть не приходится, потому что в этом случае часто происходят выкидыши. Мать нередко даже не замечает беременности, это выглядит как задержавшиеся месячные. Если мутация безвредна и встречается часто, ее называют полиморфизмом. Именно так возникли группы крови и цвета радужки. Впрочем, полиморфизм может повышать вероятность возникновения некоторых заболеваний.
Е.В. Тозлиян, педиатр-эндокринолог, генетик, к. м. н., Обособленное структурное подразделение «Научно-исследовательский клинический институт педиатрии» ГБОУ ВПО РНИМУ им. Н.И. Пирогова Минздрава РФ, г. Москва Ключевые слова
: дети, синдром Нунан, диагностика.
Key words
: children, syndrome Noonan, diagnostics.
В статье описан синдром Нунан (синдром Ульриха – Нунан, тернероидный синдром с нормальным кариотипом) – редкая врожденная патология, наследуется по аутосомно-доминантному типу, носит семейный характер, однако встречаются и спорадические случаи. Синдром предполагает наличие фенотипа, характерного для синдрома Шерешевского – Тернера у особей женского и мужского пола с нормальным кариотипом. Представлено клиническое наблюдение. Показаны сложности дифференциальнодиагностического поиска, недостаточная информированность клиницистов о данном синдроме и важность междисциплинарного подхода.
Исторические факты
Впервые о необычном синдроме упомянул О. Kobylinski в 1883 году (фото 1).
Старейший известный клинический случай синдрома Нунан, описан в 1883 году О. Kobylinski
Болезнь описана в 1963 году американским врачом-кардиологом Жаклин Нунан, сообщившей о девяти пациентах со стенозом клапана легочной артерии, малым ростом, гипертелоризмом, умеренным снижением интеллекта, птозом, крипторхизмом и скелетными нарушениями. Доктор Нунан, практиковавшая как детский кардиолог в университете Айовы, заметила, что у детей с редким типом порока сердца – стенозом клапана легочной артерии – часто наблюдались типичные физические аномалии в виде низкого роста, крыловидной шеи, широко посаженных глаз и низко расположенных ушей. Мальчики и девочки поражались одинаково. Доктор Джон Опиц, бывший студент Нунан, первым ввел в употребление термин «синдромом Нунан» для характеристики состояния детей, у которых отмечались признаки, похожие на описанные Нунан. Позже Нунан написала статью «Гипертелоризм с фенотипом Тернера», и в 1971 году на симпозиуме сердечнососудистых заболеваний название «синдром Нунан» стало официально признанным .
Этиология и патогенез
Синдром Нунан представляет собой аутосомно-доминантное заболевание с варьирующей экспрессивностью (рис. 1). Ген синдрома Нунан локализован на длинном плече хромосомы 12 . Не исключена генетическая гетерогенность синдрома. Описаны спорадические и семейные формы синдрома с аутосомно-доминантной формой наследования. В семейных случаях мутантный ген наследуется, как правило, от матери, так как из-за тяжелых пороков развития мочеполовой системы мужчины с этим заболеванием часто бесплодны. Большинство описанных случаев являются спорадическими, вызванными мутациями de novo.

. Аутосомно-доминантный тип наследования
Описанные сочетания синдрома Нунан с нейрофиброматозом I типа в нескольких семьях заставило предположить возможную связь двух независимых локусов 17q11.2 хромосомы 17. У некоторых больных выявляются микроделеции в локусе 22q11 хромосомы 22; в этих случаях клинические проявления синдрома Нунан сочетаются с гипофункцией тимуса и синдромом Ди Джорджи. Ряд авторов обсуждают участие в патогенезе синдрома предполагаемых генов лимфогенеза в связи с наличием сходных с синдромом Тернера лицевых и соматических аномалий и высокой частоты патологии лимфатической системы .
Наиболее частая причина синдрома Нунан – мутация гена PTPN11, которая обнаруживается приблизительно у 50% больных. Белок, кодируемый геном PTPN11, относится к семейству молекул, регулирующих ответ эукариотических клеток на внешние сигналы. Наибольшее число мутаций при синдроме Нунан локализовано в экзонах 3,7 и 13 гена PTPN11, кодирующих домены белка, отвечающие за переход протеина в активное состояние .
Возможные представления о патогенезе представлены следующими механизмами:
RAS-MAPK-путь – очень важный путь сигнальной трансдукции, через который внеклеточные лиганды – определенные факторы роста, цитокины и гормоны – стимулируют клеточную пролиферацию, дифференцирование, выживаемость и метаболизм (рис. 2). После связывания лиганда рецепторы на поверхности клеток фосфорилируются в местах их эндоплазматического региона. Это связывание задействует адаптерные протеины (например, GRB2), которые формируют конститутивный комплекс с факторами обмена гуаниновых нуклеотидов (например, SOS), конвертирующих неактивный ГДФ-связанный RAS в его активную ГТФ-связанную форму. Активированные RAS-протеины затем активируют RAF-MEKERKкаскад через ряд реакций фосфорилирования. В результате активированный ERK проникает в ядро для изменения транскрипции целевых генов и корректирует активность эндоплазматических мишеней для индукции адекватных кратковременных и длительных клеточных ответов на стимул. Все гены, вовлеченные в синдром Нунан, кодируют интегральные для этого пути протеины, и мутации, вызывающие болезнь, обычно усиливают сигнал, проходящий через этот путь.
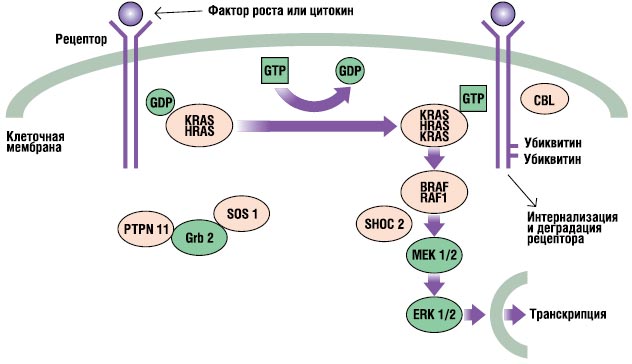
. RAS-MAPK-сигнальный путь. Ростовые сигналы передаются с активированных фактором роста рецепторов к ядру. Мутации в PTPN11, KRAS, SOS1, NRAS и RAF1 ассоциированы с синдромом Нунан, а мутации в SHOC2 и CBL ассоциированы с подобным синдрому Нунан фенотипом
Клиническая характеристика синдрома Нунан
Фенотип больных с синдромом Нунан напоминает синдром Тернера: короткая шея с крыловидной складкой или низким ростом волос, низкий рост, гипертелоризм глазных щелей (фото 2). Лицевые микроаномалии включают антимонголоидный разрез глазных щелей, опущенные вниз наружные углы глазных щелей, птоз, эпикантус, низко расположенные ушные раковины, складчатый завиток ушных раковин, аномалии прикуса, расщелину язычка мягкого неба, готическое небо, микрогнатию и микрогению. Грудная клетка щитовидной формы с гипоплазированными широко расставленными сосками, грудина выступает в верхней части и западает в нижней. Около 20% больных имеют умеренно выраженную патологию скелета. Наиболее часто встречаются воронкообразная деформация грудной клетки, кифоз, сколиоз; реже – уменьшение числа шейных позвонков и их сращение, напоминающее аномалии при синдроме Клиппеля – Фейля .

. Фенотипы синдрома Нунан
У больных с синдромом Нунан обычно светлые густые вьющиеся волосы с необычным ростом на темени, часто встречаются пигментные пятна на коже, гипертрихоз, дистрофия ногтевых пластинок, аномалии прорезывания и расположения зубов, склонность к образованию келоидных рубцов, повышенная растяжимость кожи. У трети больных отмечаются периферические лимфатические отеки, чаще лимфедема кистей и стоп проявляется у детей раннего возраста. Нередким признаком является патология зрения (миопия, косоглазие, умеренный экзофтальм и др.). Задержка роста встречается примерно у 75% больных, больше выражена у мальчиков и обычно незначительна. Отставание в росте манифестирует в первые годы жизни, реже отмечается незначительный дефицит роста и массы при рождении. С первых месяцев жизни отмечается снижение аппетита. Костный возраст обычно отстает от паспортного.
Характерным признаком синдрома является одно- или двусторонний крипторхизм, встречающийся у 70–75% больных мужского пола, у взрослых больных отмечается азооспермия, олигоспермия, дегенеративные изменения яичек. Тем не менее пубертат наступает спонтанно, иногда с некоторой задержкой. У девочек часто отмечается задержка становления менструации, иногда – нарушения менструального цикла. Фертильность может быть нормальной у больных обоих полов.
Умственная отсталость выявляется более чем у половины больных, как правило, незначительная. Часто отмечаются особенности поведения, расторможенность, синдром дефицита внимания. Речь обычно развита лучше, чем другие интеллектуальные сферы. Степень снижения интеллекта не коррелирует с тяжестью соматических нарушений [Маринчева Г.С., 1988]. В единичных случаях описываются пороки развития центральной нервной системы (гидроцефалия, спинномозговые грыжи), тромбоэмболические инфаркты мозга, возможно, связанные с гипоплазией сосудов .
Пороки внутренних органов при синдроме Нунан достаточно характерны. Наиболее типичными являются сердечно-сосудистые аномалии: клапанный стеноз легочной артерии (около 60% больных), гипертрофическая кардиомиопатия (20–30%), структурные аномалии митрального клапана, дефекты предсердной перегородки, тетрада Фалло; коарктация аорты описана только у больных мужского пола.
У трети больных регистрируются пороки мочевыделительной системы (гипоплазия почек, удвоение лоханок, гидронефроз, мегауретер и др.).
Достаточно часто при синдроме Нунан отмечается повышенная кровоточивость, особенно при оперативных вмешательствах в ротовой полости и носоглотке. Обнаруживаются различные дефекты коагуляции: недостаточность тромбоцитарной системы, снижение уровня факторов свертывания, особенно XI и XII, увеличение тромбопластинового времени . Имеются сообщения о сочетании синдрома Нунан с лейкемией и рабдомиосаркомой, что может свидетельствовать о некотором повышении риска малигнизации у этих больных .
В таблице 1 представлены особенности фенотипа при синдроме Нунан, меняющиеся с возрастом пациента. В таблице 2 – корреляция между фенотипом и генотипом при синдроме Нунан.
Таблица 1 . Типичные черты лица у больных синдромом Нунан по возрастам
| Лоб, лицо, волосы | Глаза | Уши | Нос | Рот | Шея | |
| Новорожденный* | Высокий лоб, низкая линия роста волос в затылочной области | Гипертелоризм, наклонные книзу глазные щели, складка эпикантуса | – | Короткий и широкий утопленный корень, вздернутый кончик | Глубоко утопленный губной желобок, высокие широкие пики красной каймы губ, микрогнатия | Избыточная кожа на затылке |
| Грудной (2–12 мес.) | Большая голова, высокий и выпирающий лоб | Гипертелоризм, птоз или толстые нависающие веки | – | Короткий и широкий утопленный корень | – | – |
| Ребенок (1–12 лет) | Грубые черты, вытянутое лицо | – | – | – | – | – |
| Подросток (12–18 лет) | Миопатичес-кое лицо | – | – | Мостик высокий и тонкий | – | Очевидное формирование шейных складок |
| Взрослый (>18 лет) | Отличительные черты лица утонченные, кожа кажется тонкой и прозрачной | – | – | Выпирающая носогубная складка | – | – |
| Все возрасты | – | Голубые и зеленые радужные оболочки, ромбовидные брови | Низкие, ротированные назад уши с толстыми складками | – | – | – |
Таблица 2 . Корреляции между генотипом и фенотипом при синдроме Нунан*
| Сердечнососудистая система | Рост | Развитие | Кожа и волосы | Другое | |
| PTPN11 (примерно 50%) | Более выражен стеноз легочного ствола; меньше – гипертрофическая кардиомиопатия и дефект межпредсердной перегородки | Более низкий рост; ниже концентрация IGF1 | Пациенты с N308D и N308S имеют слабое снижение или нормальный интеллект | – | Больше выражен геморрагический диатез и ювенильная миеломоноцитарная лейкемия |
| SOS1 (примерно 10%) | Меньше дефект межпредсердной перегородки | Более высокий рост | Меньше снижение интеллекта, задержка развития речи | Подобны сердечно-кожно-лицевому синдрому | – |
| RAF1 (примерно 10%) | Больше тяжелая гипертрофическая кардио-миопатия | – | – | Больше родимых пятен, лентиго, пятен кофе с молоком | – |
| KRAS (<2%) | – | – | Более тяжелая задержка когнитивного развития | Подобны сер-дечно-кожно-лицевому синдрому | – |
| NRAS (<1%) | – | – | – | – | – |
Данные лабораторных и функциональных исследований
Специфических биохимических маркеров для диагностики синдрома Нунан не существует. У некоторых больных выявляется снижение спонтанной ночной секреции гормона роста при нормальном ответе на фармакологические стимулирующие тесты (клофелином и аргинином), снижение уровня соматомедина-С и снижение реакции соматомединов на введение гормона роста.
Критерии диагноза
Диагноз «синдром Нунан» ставится на основании клинических признаков, в некоторых случаях диагноз подтверждается результатами молекулярно-генетического исследования. Критерии диагностики синдрома включают наличие характерного лица (при нормальном кариотипе) в сочетании с одним из следующих признаков: патологии сердца, низкий рост или крипторхизм (у мальчиков), задержка полового созревания (у девочек). Для выявления сердечно-сосудистой патологии необходимо проведение ультразвукового исследования сердца с динамическим определением размеров полостей и стенки желудочков. Возможна пренатальная диагностика заболевания при помощи ультразвукового мониторинга, позволяющего выявить пороки сердца и аномалии строения шеи .
Дифференциальная диагностика
У девочек дифференциальный диагноз проводится в первую очередь с синдромом Тернера; уточнить диагноз позволяет цитогенетическое исследование. Фенотипические признаки синдрома Нунан встречаются при ряде других заболеваний: синдроме Вильямса, синдроме LEOPARD, Дубовица, кардиофацио-кожном синдроме, Корнелии де Ланге, Коэна, Рубинштейна – Тейби и др. Точная идентификация этих заболеваний будет возможна только при проведении молекулярногенетических исследований каждого синдрома при значительном клиническом материале, что в настоящее время активно развивается.
Лечение
Лечение больных с синдромом Нунан направлено на устранение пороков сердечно-сосудистой системы, нормализацию психических функций, стимуляцию роста и полового развития. Для лечения больных с дисплазией клапанов легочной артерии, помимо прочих методов, с успехом применяется баллонная вальвулопластика. С целью стимуляции психического развития применяются ноотропные и сосудистые средства. Препараты, направленные на стимуляцию полового развития, показаны в основном больным с крипторхизмом. Применяются препараты хорионического гонадотропина в возрастных дозировках. В старшем возрасте – при наличии гипогонадизма – препараты тестостерона. В последние годы применяются рекомбинантные формы гормона роста человека в лечении больных с синдромом Нунан . Клинические данные подтверждаются увеличением на фоне терапии уровня соматомедина-С и специфического связывающего белка. Конечный рост больных, длительное время получающих терапию гормоном роста, в некоторых случаях превышает средний рост членов семьи.
Прогноз для жизни определяется тяжестью сердечно-сосудистой патологии.
Профилактика болезни основывается на данных медико-генетического консультирования.
Медико-генетическое консультирование
При медико-генетическом консультировании следует исходить из аутосомно-доминантного типа наследования и высокого (50%) риска повторения заболевания в семье при унаследованных формах. С целью идентификации характера типа наследования необходимо проводить тщательное обследование родителей, так как синдром может проявляться минимальными клиническими симптомами. В настоящее время разработана и совершенствуется молекулярно-генетическая диагностика заболевания путем типирования мутаций в генах: PTPN11, SOS1, RAF1, KRAS, NRAS и др. Разрабатываются способы пренатальной диагностики заболевания.
Клиническое наблюдение
Мальчик Г., 9 лет (фото 3), наблюдался по месту жительства врачом-генетиком с диагнозом «хромосомная патология?, синдром Вильямса (своеобразный фенотип, уплотнение створок митрального клапана, гиперкальциемия однократно в 3 года)?.

. Особенности фенотипа ребенка с синдромом Нунан (удлиненный лицевой скелет с «пухлыми щечками», короткая шея, крыловидные складки на шее, нос укорочен с открытыми вперед ноздрями, пухлые губы, скошенный подбородок, антимонголоидный разрез глазных щелей, неправильный прикус, макростомия)
Жалобы на сниженную память, утомляемость, сниженные темпы роста.
Анамнез семейный : родители русские по национальности, не состоящие в кровном родстве и не имеющие профессиональных вредностей, здоровые. Рост отца – 192 см, рост матери – 172 см. В родословной случаев психических заболеваний, эпилепсии, задержки в развитии не отмечалось.
Анамнез жизни и заболевания : мальчик от 2-й беременности (1-я беременность – м/а), протекавшей с угрозой прерывания на всем протяжении, сопровождающейся многоводием. Роды первые, в срок, стремительные, масса при рождении – 3400 г, длина – 50 см. Закричал сразу, оценка по шкале Апгар – 7/9 баллов. При рождении неонатологом обращено внимание на необычный фенотип ребенка, рекомендовано исследование кариотипа, результат – 46, XY (нормальный мужской кариотип). Был заподозрен врожденный гипотиреоз, проведено исследование тиреоидного профиля, результат – нормальный тиреоидный статус. Далее ребенок наблюдался генетиком с предполагаемым диагнозом «синдром Вильямса». Ранний постнатальный период – без особенностей. Моторное развитие по возрасту, первые слова – к году, фразовая речь – в 2 года 3 мес.
В возрасте 8 лет консультирован эндокринологом по поводу сниженных темпов роста, утомляемости, сниженной памяти. При рентгенологическом исследовании кистей рук выявлено умеренное отставание костного возраста (КВ) от паспортного (КВ соответствовал 6 годам). При исследовании тиреоидного профиля выявлено умеренное повышение тиреотропного гормона при нормальном уровне свободного Т4 и остальных показателей; УЗИ щитовидной железы – без патологии. Назначена гормональная терапия с последующим динамическим наблюдением.
Учитывая неопределенность диагноза по месту жительства, генетиком ребенок направлен в Московский областной консультативно-диагностический центр для детей с целью уточнения диагноза.
Данные объективного исследования:
Рост – 126 см, вес – 21 кг.
Физическое развитие ниже среднего, гармоничное. Sds роста соответствует –1 (норма – –2+2). Особенности фенотипа (фото 3): удлиненный лицевой скелет с «пухлыми щечками», короткая шея, крыловидные складки на шее, низкий рост волос на шее, нос укороченный с открытыми вперед ноздрями, пухлые губы, скошенный подбородок, антимонголоидный разрез глазных щелей, неправильный прикус, макростомия, гипертелоризм сосков, асимметрия грудной клетки, на стопах неполная кожная синдактилия 2–3-го пальцев, выраженная гипермобильность межфаланговых суставов, ломкие, сухие ногти. По внутренним органам – без особенностей. Половое развитие – Tanner I (что соответствует допубертатному периоду).
Данные лабораторных и функциональных исследований:
Клинический анализ крови и мочи – норма.
Биохимический анализ крови – показатели в пределах нормы.
Тиреоидный профиль (ТТГ) – 7,5 мкМЕ/ мл (норма – 0,4–4,0), остальные показатели в норме.
Соматотропный гормон (СТГ) – 7 нг/мл (норма – 7–10), соматомедин-С – 250 нг/мл (норма – 88–360).
УЗИ щитовидной железы – без патологии.
УЗИ внутренних органов – без особенностей.
ЭКГ – синусовая тахикардия, нормальное положение электрической оси сердца.
ЭхоКГ – ПМК I степени с минимальной регургитацией, миксоматозное утолщение створок митрального клапана, дополнительная хорда в полости левого желудочка.
R-графия позвоночника – правосторонний сколиоз грудного отдела позвоночника I степени.
R-графия кистей рук с захватом предплечий – костный возраст 7–8 лет.
ЭЭГ-паттернов эпилептической активности не зарегистрировано.
МРТ головного мозга – без патологических изменений.
Аудиограмма – без патологии.
ДНК-диагностика: молекулярно-генетическое исследование – делеций исследуемых локусов критического района хромосомы 7 не выявлено; обнаружена мутация Gly434Ary (1230G>A) в 11-м экзоне гена SOS1 (анализ гена PTPN11 – мутаций не обнаружено), что характерно для синдрома Нунан.
Консультации специалистов:
Эндокринолог – субклинический гипотиреоз, неполная медикаментозная компенсация.
Окулист – астигматизм.
Невролог – вегетососудистая дистония. Невротические реакции.
Кардиолог – функциональная кардиопатия.
Хирург-ортопед – нарушение осанки. Деформация грудной клетки.
Генетик – синдром Нунан.
Учитывая фенотип ребенка, данные анамнеза, результаты дополнительных исследований, поставлен диагноз «синдром Нунан», что подтверждено результатом молекулярно-генетического исследования.
Таким образом, представленное клиническое наблюдение демонстрирует сложности дифференциально-диагностического поиска, необходимость интегрировать отдельные признаки в общий фенотип того или иного патологического состояния для целенаправленной своевременной диагностики отдельных форм наследственных заболеваний, важность молекулярно-генетических методов для уточнения диагноза. Своевременная диагностика, уточнение генеза каждого синдрома особенно важны, так как позволяют найти оптимальный подход к лечению этих состояний, профилактике возможных осложнений (вплоть до инвалидности ребенка); предупреждению повторного возникновения наследственных болезней в пораженных семьях (медико-генетическое консультирование). Это диктует необходимость врачам различных специальностей четко ориентироваться в потоке наследственно обусловленной патологии.
Список литературы:
- Baird P., De Jong B. Noonan’s syndrome (XX and XY Turner phenotype) in three generations of a family // J. Pediatr., 1972, vol. 80, p. 110–114.
- Hasegawa T., Ogata T. et al. Coarctation of the aorta and renal hupoplasia in a boy with Turner/Noonan surface anomalies and a 46, XY karyotype: a clinical model for the possible impairment of a putative lymphogenic gene(s) for Turner somatic stigmata // Hum. Genet., 1996, vol. 97, р. 564–567.
- Федотова Т.В., Кадникова В.А. и соавт. Клинико-молекулярно-генетический анализ синдрома Нунан. Материалы VI съезда Российского общества медицинских генетиков. Медицинская генетика, приложение к № 5, 2010, с.184.
- Ward K.A., Moss C., McKeown C. The cardio-facio-cutaneous syndrome: a manifestation of the Noonan syndrome? // Br. J. Dermatol., 1994, vol. 131, р. 270–274.
- Municchi G., Pasquino A.M. et al. Growth hormone treatment in Noonan syndrome: report of four cases who reached fi nal height // Horm. Res., 1995, vol. 44, р. 164–167.
Трем группам американских ученых, независимо друг от друга, удалось впервые установить связь между мутациями в определенных генах и вероятностью развития у ребенка расстройств аутистического спектра, сообщает The New York Times. Кроме того, исследователи нашли научное подтверждение ранее выявленной прямой зависимости между возрастом родителей, в особенности отцов, и риском развития аутизма у потомства.
Все три группы сосредоточились на редко встречающейся группе генетических мутаций, названной "de novo". Эти мутации не наследуются, а возникают в процессе зачатия. В качестве генетического материала были взяты образцы крови членов семей, в которых родители не являлись аутистами, а у детей развились различные расстройства аутистического спектра.
Первая группа ученых под руководством Мэтью Стэйта (Matthew W. State), профессора генетики и детской психиатрии из Йэльского университета, чья работа опубликована 4 апреля в журнале Nature, проанализировала наличие мутаций "de novo" у 200 человек с диагнозом "аутизм", чьи родители, братья и сестры не являлись аутистами. В результате были обнаружены два ребенка с одинаковой мутацией в одном и том же гене, при этом их не связывало больше ничего, кроме диагноза.
"Это как при игре дартс дважды попасть дротиком в одну и ту же точку на мишени. Вероятность того, что обнаруженная мутация связана с аутизмом - 99,9999 процентов", - цитирует издание профессора Стэйта.
Команда под руководством Ивэна Эйхлера (Evan E. Eichler), профессора генетики из университета штата Вашингтон, исследуя образцы крови 209 семей, в которых есть дети-аутисты, обнаружила такую же мутацию в том же самом гене у одного ребенка. Кроме того, были выявлены двое детей-аутистов из разных семей, у которых оказались идентичные между собой мутации "de novo", но в других генах. Таких совпадений у испытуемых - не аутистов замечено не было.
Третья группа исследователей, которую возглавлял профессор Марк Дэли (Mark J. Daly) из Гарвардского университета, обнаружила у детей-аутистов несколько случаев мутаций типа "de novo" в тех же трех генах. Хотя бы одна мутация такого типа присутствует в генотипе любого человека, но, полагает Дэли, у аутистов, в среднем, их значительно больше.
Все три группы исследователей также подтвердили и ранее замеченную связь между возрастом родителей и аутизмом у ребенка. Чем старше родители, в первую очередь отец, тем выше риск появления мутаций "de novo". Проанализировав 51 мутацию, команда под руководством профессора Эйхлера выяснила, что в мужской ДНК такого рода повреждения встречаются в четыре раза чаще, чем в женской. И тем чаще, если возраст мужчины превышает 35 лет. Таким образом, предполагают ученые, именно поврежденный отцовский генетический материал, получаемый потомством при зачатии, является источником тех мутаций, которые влекут за собой развитие аутистических расстройств.
Ученые согласны в том, что поиск путей предотвращения такого развития событий будет долгим, исследование генетической природы аутизма только начинается. В частности, команды Эйхлера и Дэли нашли свидетельства того, что гены, в которых обнаружены мутации "de novo", задействованы в одних и тех же биологических процессах. "Но это лишь верхушка верхушки айсберга, - считает профессор Эйхлер. - Главное, что все мы сошлись на том, с чего надо начать".
Синдром возникает из-за отсутсвия части генетического материала расположенного на коротком плече 11 хромосомы. Удаление части генетического материала называют делецией. Делеция приводит к поражению тех функций, которые должны были выполняться утраченными генами.
Все гены, за исключением некоторых, которые расположены в половых хромосомах представлены в двойном экземпляре. Одну порцию генов каждый человек получает от матери, вторую идентичную от отца. Те в свою очередь получили свои пары генов от своих родителей. Генетический материал передается от родителей через половые клетки. Половые клетки (яйцеклетка или сперматозоид) это единственные клетки в организме, которые несут только одну копию генетического материала. Перед тем как генетический материал попадает в половую клетку, между двумя копиями генов идет перетасовка генов и в каждый родитель помещает в половую клетку генетический материал который представляет собой микст от того материала который он в свою очередь получил от своих родителей. Новая жизнь их тоже будет тасовать перед тем как поместить в половую клетку для создания следующего поколения. Этот процесс называется кроссинговер. Он происходит между гомологичными участками хромосом, в процессе формирования половых клеток. В процессе кроссинговера гены могут создавать новые комбинации. Такое смешивание обеспечивает разнообразие новых поколений. Для чего это нужно? Это нужно для того, чтобы обеспечить изменчивость поколения, в противном случае мы бы передавали нашим детям точные копии хромосом полученными от одного из наших родителей, изменчивость поколений была бы крайне ограничена, что сделало бы биологическую эволюцию на Земле крайне затруднительной, а следовательно и уменьшило бы шансы на выживание. В момент когда проходят такие процессы может оторваться кусочек хромосомы и получиться “делеция”. Делеция это вид мутации. Если возникла впервые, то такая мутация называется мутацией de novo (самая первая, начальная). Кроме мутаций, которые впервые возникли в организме, существуют мутации, которые передались по наследству. Мутация de novo может быть передана следующим поколениям, тогда она уже не будет называться мутацией de novo.
При WAGR синдроме часть генетического кода удалена и генетического материала не хватает.
В природе существуют обратные состояния, когда заболевание проявляется из-за лишней копии генетического материала.
Проявление WAGR синдрома зависит от того какие именно гены выключены в результате делеции. Всегда выпадают соседние гены. При WAGR всегда выпадают PAX6 ген и WT1 ген, что ведет к типичному проявлению заболевания. Точечные мутации гена PAX6 ведут к аниридии, а мутации WT1 ведут к возникновению опухоли Вильмса. При WAGR нет мутации этих генов - отсутствуют сами гены.
У людей с WAGRO (добавилась буква О - obesity) синдромом есть поражение гена BDNF. Этот ген экспрессируется в головном мозгу и важен в жизни нейронов. Протеин, который продуцируется под влиянием этого гена, скорее всего участвует в регуляции насыщения, жажды и массы тела. Потеря BDNF скорее всего связана с ожирением, которое начинается в детском возрасте у детей с WAGRO синдромом. Пациенты с WAGRO имеют больший риск неврологических проблем таких как снижение интеллекта, аутизм. Не изучено до конца, связан ли этот риск именно с потерей гена BDNF
Про гены, которые выключились при WAGR синдроме мы кое что знаем:
WT1
WT1 - ген (Wilms tumor gene), который секретирует протеин, необходимый для нормального развития почек и гонад (яичников у женщин и яичек у мужчин). В этих тканях протеин играет роль в дифференциации клеток и апоптоза. Для проведения всех этих функция WT1 регулирует активность других генов путем связывания регионов ДНК.
Ген WT1 необходим для подавления опухоли Вильмса. Встречается вариант названия гена Wilm"s tumour tumor suppressor gene1 (ген подавляющий развития опухоли Вильмса). Его мутация или отсутствие ведет к увеличенному риску развития опухоли. Именно из-за вероятности вовлечения этого гена в WAGR синдром необходим перманентный контроль за состоянием почек.
PAX6
PAX6 относится к семейству генов, которые играют критическую роль в развитии органов и тканей во время эмбрионального развития. Члены семейства PAX важны для нормального функционирования разных клеток организма после рождения. Гены семейства PAX участвуют в синтезе протеинов, которые связывают специфические участки ДНК и так контролируют активность других генов. Из-за такого свойства, PAX протеины называют факторами транскрипции (transcription factors)
В период эмбрионального развития, PAX 6 белок активирует гены вовлеченные в развитие глаз, мозга, спинного мозга и поджелудочной железы. PAX 6 участвует в развитии нервных клеток ольфакторного тракта, которые отвечают за обоняние. В настоящее время функция PAX 6 во время внутриутробного развития скорее всего не изучена до конца и со временем мы получаем новые факты. После рождения PAX6 протеин регулирует множество генов в глазу.
Недостаточность функции гена PAX 6 ведет к тому, что проблемы с глазами возникают после рождения.
BDNF
BDNF ген кодирует белок, который обнаруживается в головном мозге и в спинном мозге. Этот ген играет роль в росте, созревании нервных клеток. BDNF белок активен в синапсах головного мозга. Синапсы могут изменяться и адаптироваться в ответ на опыт. BDNF белок помогает регулировать изменчивость синапсов, что очень важно для процессов обучения и памяти.
BDNF протеин найден в регионах головного мозга, которые отвечают за сытость, жажду и вес тела. Скорее всего этот белок вносит вклад в эти процессы.
Экспрессия этого гена снижена при болезнях Альцгеймера, Паркинсона и Хантингтона, этот ген может играть роль в ответах на стресс и в болезнях расстройства настроения. Ген BDNF привлекает внимания многих исследователей. Существуют работы, которые изучают активность белка BDNF в головном мозгу в зависимости от физических упражнений, диеты, умственного напряжения и других состояний. Активность данного белка связывают с ментальной деятельностью и психическими состояниями, проводятся попытки влиять на его уровень.
Я буду благодарен за указание мне о новой информации по этому вопросу. Пишите все в комментариях.
Примечание:
Слова протеин и белок являются синонимами
ГЛАВА 5 ИЗМЕНЧИВОСТЬ ОРГАНИЗМА
ГЛАВА 5 ИЗМЕНЧИВОСТЬ ОРГАНИЗМА
Общие данные
Изменчивость организма есть изменчивость его генома, обусловливающая генотипические и фенотипические различия человека и вызывающая эволюционное разнообразие его генотипов и фенотипов (см. главы 2 и 3).
Внутриутробное развитие зародыша, эмбриона, плода, дальнейшее постнатальное развитие организма человека (младенчество, детство, отрочество, юность, взрослая жизнь, старение и смерть) осуществляются в соответствии с генетической программой онтогенеза, сформировавшейся при слиянии материнского и отцовского геномов (см. главы 2 и 12).
В ходе онтогенеза геном организма индивида и закодированная в нем информация подвергаются непрерывным преобразованиям под действием факторов окружающей среды. Возникшие в геноме изменения могут передаваться из поколения в поколение, обусловливая изменчивость признаков и фенотипа организма у потомков.
В начале XX в. немецкий зоолог В. Хэкер выделил направление генетики, посвященное изучению связей и взаимоотношений между генотипами и фенотипами и анализу их изменчивости, и назвал его феногенетикой.
В настоящее время феногенетики выделяют два класса изменчивости: ненаследственную (или модификационную), которая не передается из поколения в поколение, и наследственную, которая передается из поколения в поколение.
В свою очередь, наследственная изменчивость также бывает двух классов: комбинативная (рекомбинационная) и мутационная. Изменчивость первого класса определяют три механизма: случайные встречи гамет при оплодотворении; кроссинговер, или мейотическая рекомбинация (обмен равными участками между гомологичными хромосомами в профазе первого деления мейоза); независимое расхождение гомологичных хромосом к полюсам деления при образовании дочерних клеток в ходе митоза и мейоза. Изменчивость второго
класса обусловлена точковыми, хромосомными и геномными мутациями (см. ниже).
Последовательно рассмотрим различные классы и типы изменчивости организма на разных этапах его индивидуального развития.
Изменчивость при оплодотворении гамет и начало функционирования генома зародившегося организма
Материнский и отцовский геномы не могут функционировать отдельно друг от друга.
Только двух родительских генома, объединившиеся в зиготе, обеспечивают зарождение молекулярной жизни, появление нового качественного состояния - одного из свойств биологической материи.
На рис. 23 отражены результаты взаимодействия двух родительских геномов при оплодотворении гамет.
Согласно формуле оплодотворения: зигота = яйцеклетка + сперматозоид, начало развития зиготы - это момент формирования двойного (диплоидного) при встрече двух гаплоидных наборов родительских гамет. Именно тогда зарождается молекулярная жизнь и запускается цепь последовательных реакций на основе сначала экспрессии генов генотипа зиготы, а затем генотипов появившихся из нее дочерних соматических клеток. Отдельные гены и группы генов в составе генотипов всех клеток организма начинают «включаться» и «выключаться» в ходе реализации генетической программы онтогенеза.
Ведущая роль в происходящих событиях принадлежит яйцеклетке, имеющей в ядре и цитоплазме все необходимые для зарожде-
Рис. 23. Результаты взаимодействия двух родительских геномов при оплодотворении гамет (рисунки по www.bio.1september.ru; www.bio.fizteh.ru; www. vetfac.nsau.edu.ru соответственно)
ния и продолжения жизни структурные и функциональные компоненты ядра и цитоплазмы (суть биологического матриархата). Сперматозоид же содержит ДНК и не содержит компонентов цитоплазмы. Проникнув в яйцеклетку, ДНК сперматозоида вступает в контакт с ее ДНК, и тем самым в зиготе «включается» функционирующий в течение всей жизни организма главный молекулярный механизм: ДНК-ДНК взаимодействие двух родительских геномов. Строго говоря, активизируется генотип, представленный примерно равными частями нуклеотидных последовательностей ДНК материнского и отцовского происхождения (без учета мтДНК цитоплазмы). Упростим сказанное: начало молекулярной жизни в зиготе - нарушение постоянства внутренней среды яйцеклетки (ее гомеостаза), а вся последующая молекулярная жизнь многоклеточного организма - стремление восстановить подверженный действию факторов среды гомеостаз или баланс между двумя противоположными состояниями: стабильностью с одной стороны и изменчивостью с другой. Таковы причинно-следственные связи, определяющие возникновение и непрерывность молекулярной жизни организма в ходе онтогенеза.
Теперь обратим внимание на результаты и значение изменчивости генома организма как продукта эволюции. Сначала рассмотрим вопрос об уникальности генотипа зиготы или клетки-родоначальницы всех клеток, тканей, органов и систем организма.
Само оплодотворение происходит случайно: одну женскую гамету оплодотворяет только одна мужская гамета из 200-300 млн сперматозоидов, содержащихся в эякуляте мужчины. Очевидно, что каждую яйцеклетку и каждый сперматозоид отличают друг от друга многие генотипические и фенотипические признаки: наличие измененных или неизмененных по составу и комбинациям генов (результаты комбинативной изменчивости), разные сиквенсы нуклеотидных последовательностей ДНК, разные размеры, форма, функциональная активность (подвижность), зрелость гамет и др. Именно эти отличия позволяют говорить об уникальности генома любой гаметы и, следовательно, генотипа зиготы и всего организма: случайность оплодотворения гамет обеспечивает появление на свет генетически уникального организма индивида.
Иными словами, молекулярная жизнь человека (как и жизнь биологического существа вообще) - «дар судьбы» или, если угодно, «божественный дар», ибо вместо данного индивида с одинаковой
вероятностью могли родиться генетически иные - его родные братья и сестры.
Теперь продолжим наши рассуждения о балансе между стабильностью и изменчивостью наследственного материала. В широком смысле, поддержание такого баланса - это одновременное сохранение и изменение (преобразование) стабильности наследственного материала под действием внутренних (гомеостаз) и внешних средовых факторов (норма реакции). Гомеостаз зависит от генотипа, обусловленного слиянием двух геномов (см. рис. 23). Норма реакции определяется взаимодействием генотипа с факторами окружающей среды.
Норма и диапазон реакции
Специфический способ реакции организма в ответ на действие факторов окружающей среды называется нормой реакции. Именно гены и генотип ответственны за развитие и диапазон модификаций отдельных признаков и фенотипа всего организма. Вместе с тем, в фенотипе реализуются далеко не все возможности генотипа, т.е. фенотип - частный (для индивида) случай реализации генотипа в конкретных условиях окружающей среды. Поэтому, например, между монозиготными близнецами, имеющими полностью идентичные генотипы (100% общих генов), выявляются заметные фенотипические различия, если близнецы растут в разных условиях окружающей среды.
Норма реакции бывает узкой или широкой. В первом случае стабильность отдельного признака (фенотипа) сохраняется практически вне зависимости от влияния окружающей среды. Примерами генов с узкой нормой реакции или непластичных генов служат гены, кодирующие синтез антигенов групп крови, окраску глаз, курчавость волос и др. Их действие одинаково при любых (совместимых с жизнью) внешних условиях. Во втором случае стабильность отдельного признака (фенотипа) изменяется в зависимости от влияния окружающей среды. Пример генов с широкой нормой реакции или пластичных генов - гены, контролирующие количество эритроцитов крови (разное у лиц, поднимающихся в гору, и лиц, спускающихся с горы). Другой пример широкой нормы реакции - изменение окраски кожных покровов (загар), связанный с интенсивностью и временем воздействия на организм ультрафиолетового облучения.
Говоря о диапазоне реакции, следует иметь в виду фенотипические различия, проявляющиеся у индивида (его генотипа) в зависимости от
«обедненных» или «обогащенных» условий окружающей среды, в которых находится организм. Согласно определению И.И. Шмальгаузена (1946), «наследуются не признаки, как таковые, а норма их реакции на изменения условий существования организмов».
Таким образом, норма и диапазон реакции - это пределы генотипической и фенотипической изменчивости организма при изменении условий окружающей среды.
Следует также отметить, что из внутренних факторов, оказывающих влияние на фенотипическое проявление генов и генотипа, определенное значение имеют пол и возраст индивида.
Внешние и внутренние факторы, определяющие развитие признаков и фенотипов, входят в указанные в главе три группы основных факторов, среди которых гены и генотип, механизмы межмолекулярных (ДНК-ДНКовых) и межгенных взаимодействий между родительскими геномами и факторы окружающей среды.
Безусловно, основой приспособления организма к условиям окружающей среды (основой онтогенеза) является его генотип. В частности, индивиды с генотипами, не обеспечивающими подавление отрицательного действия патологических генов и факторов среды, оставляют меньше потомков, чем те индивиды, у которых нежелательные эффекты подавляются.
Вероятно, что в генотипы более жизнеспособных организмов включены специальные гены (гены-модификаторы), подавляющие действие «вредных» генов таким образом, что вместо них доминантными становятся аллели нормального типа.
НЕНАСЛЕДСТВЕННАЯ ИЗМЕНЧИВОСТЬ
Говоря о ненаследственной изменчивости генетического материала, снова рассмотрим пример широкой нормы реакции - изменение окраски кожных покровов под действием ультрафиолетового излучения. «Загар» ведь не передается из поколения в поколение, т.е. не наследуется, хотя в его возникновении участвуют пластичные гены.
Точно так же не наследуются результаты травм, рубцовые изменения тканей и слизистых оболочек при ожоговой болезни, обморожениях, отравлениях и многие другие признаки, вызванные действием исключительно факторов среды. Вместе с тем, следует подчеркнуть: ненаследственные изменения или модификации связаны с наслед-
ственными свойствами данного организма, ибо образуются на фоне конкретного генотипа в конкретных условиях окружающей среды.
Наследственная комбинативная изменчивость
Как сказано в начале главы, кроме механизма случайных встреч гамет при оплодотворении, комбинативная изменчивость включает механизмы кроссинговера в первом делении мейоза и независимого расхождения хромосом к полюсам деления при образовании дочерних клеток в ходе митоза и мейоза (см. главу 9).
Кроссинговер в первом делении мейоза
За счет механизма кроссинговера сцепление генов с хромосомой регулярно нарушается в профазе первого деления мейоза в результате перемешивания между собой (обмена) генов отцовского и материнского происхождения (рис. 24).
В начале XX в. при открытии кроссинговера Т.Х. Морган и его ученики предположили: кроссинговер между двумя генами может происходить не только в одной, но и в двух, трех (соответственно двойной и тройной кроссинговер) и большем количестве точек. Отмечалось подавление кроссинговера в участках, непосредственно примыкающих к точкам обмена; такое подавление назвали интерференцией.
В конечном итоге подсчитали: на один мужской мейоз приходится от 39 до 64 хиазм или рекомбинаций, а на один женский мейоз - до 100 хиазм.
 Рис. 24.
Схема кроссинговера в первом делении мейоза (по Шевченко В.А. и соавт., 2004):
Рис. 24.
Схема кроссинговера в первом делении мейоза (по Шевченко В.А. и соавт., 2004):
a - сестринские хроматиды гомологичных хромосом до начала мейоза; б - они же во время пахитены (видна их спирализация); в - они же во время диплотены и диакинеза (стрелки указывают на места кроссинговера-хиазмы, или участки обмена)
В результате сделали вывод: сцепление генов с хромосомами постоянно нарушается в ходе кроссинговера.
Факторы, влияющие на кроссинговер
Кроссинговер - один из регулярных генетических процессов в организме, контролируемый многими генами как непосредственно, так и через физиологическое состояние клеток в ходе мейоза и даже митоза.
К факторам, влияющим на кроссинговер, относятся:
Гомо- и гетерогаметный пол (речь идет о митотическом кроссинговере у самцов и самок таких эукариот, как дрозофила и тутовый шелкопряд); так, у дрозофилы кроссинговер протекает нормально; у тутового шелкопряда - либо тоже нормально, либо отсутствует; у человека следует обратить внимание на смешанный («третий») пол и конкретно на роль кроссинговера при аномалиях развития пола при мужском и женском гермафродитизме (см. главу 16);
Структура хроматина; на частоту кроссинговера в разных участках хромосом влияет распределение гетерохроматиновых (прицентромерные и теломерные участки) и эухроматиновых районов; в частности, в прицентромерных и теломерных участках частота кроссинговера снижена, и расстояние между генами, определяемое по частоте кроссинговера, может не соответствовать фактическому;
Функциональное состояние организма; по мере увеличения возраста меняется степень спирализации хромосом и скорость клеточного деления;
Генотип; в его составе выделены гены, увеличивающие или уменьшающие частоту кроссинговера; «запиратели» последнего - хромосомные перестройки (инверсии и транслокации), затрудняющие нормальную конъюгацию хромосом в зиготене;
Экзогенные факторы: воздействие температуры, ионизирующей радиации и концентрированных растворов солей, химические мутагены, лекарства и гормоны, как правило, повышающие частоту кроссинговера.
По частоте мейотического и митотического кроссинговера и СХО порой судят о мутагенном действии лекарств, канцерогенов, антибиотиков и других химических соединений.
Неравный кроссинговер
В редких случаях в ходе кроссинговера наблюдаются разрывы в несимметричных точках сестринских хроматид, и они обменива-
ются между собой неравными участками - это неравный кроссинговер.
Вместе с тем, описаны случаи, когда в ходе митоза наблюдается митотическая конъюгация (неправильное спаривание) гомологичных хромосом и рекомбинация происходит между несестринскими хроматидами. Такое явление получило название генной конверсии.
Значение данного механизма трудно переоценить. Например, в результате неправильного спаривания гомологичных хромосом по фланкирующим повторам может произойти удвоение (дупликация) или утрата (делеция) участка хромосомы, содержащего ген РМР22, что обусловит развитие наследственной аутосомно-доминантной моторно-сенсорной нейропатии Шарко-Мари-Тус.
Неравный кроссинговер - один из механизмов возникновения мутаций. Например, периферический белок миелин кодируется геном РМР22, расположенным в хромосоме 17 и имеющим длину около 1,5 млн н.п. Этот ген фланкируется двумя гомологичными повторами длиной около 30 тыс. н.п. (повторы расположены на флангах гена).
Особенно много мутаций в результате неравного кроссинговера происходит в псевдогенах. Тогда либо фрагмент одного аллеля переносится в другой аллель, либо фрагмент псевдогена - в ген. Например, подобная мутация отмечается при переносе последовательности псевдогена в ген 21-гидроксилазы (CYP21B) при адреногенитальном синдроме или врожденной гиперплазии коры надпочечников (см. главы 14 и 22).
Кроме того, за счет рекомбинаций в ходе неравного кроссинговера могут образовываться множественные аллельные формы генов, кодирующих антигены HLA класса I.
Независимое расхождение гомологических хромосом к полюсам деления при образовании дочерних клеток в ходе митоза и мейоза
Благодаря процессу репликации, предшествующему митозу соматической клетки, общее количество нуклеотидных последовательностей ДНК увеличивается вдвое. Формирование одной пары гомологичных хромосом происходит из двух отцовских и двух материнских хромосом. При распределении этих четырех хромосом в две дочерние клетки каждая из клеток получит одну отцовскую и одну материнскую хромосомы (для каждой пары хромосомного набора), однако какую именно из двух, первую или вторую, неизвестно. Имеет место
случайный характер распределения гомологичных хромосом. Легко подсчитать: за счет различных комбинаций 23 пар хромосом общее количество дочерних клеток составит 2 23 , или более 8 млн (8 χ 10 6) вариантов комбинаций хромосом и расположенных на них генов. Следовательно, при случайном характере распределения хромосом в дочерние клетки каждая из них будет иметь свой уникальный кариотип и генотип (свой вариант комбинации хромосом и сцепленных с ними генов соответственно). Следует отметить и возможность патологического варианта распределения хромосом в дочерние клетки. Например, попадание в одну из двух дочерних клеток только одной (отцовской или материнской по происхождению) Х-хромосомы приведет к моносомии (синдром Шерешевского-Тернера, кариотип 45, ХО), попадание трех одинаковых аутосом приведет к трисомии (синдромы Дауна, 47,XY,+21; Патау, 47,ХХ,+13 и Эдвадса, 47,ХХ,+18; см. также главу 2).
Как отмечено в главе 5, в одну дочернюю клетку могут одновременно попасть две отцовские или две материнские по происхождению хромосомы - это однородительская изодисомия по конкретной паре хромосом: синдромы Сильвера-Рассела (две материнские хромосомы 7), Беквитта-Видемана (две отцовские хромосомы 11), Ангельмана (две отцовские хромосомы 15), Прадера-Вилли (две материнские хромосомы 15). В целом объем нарушений распределения хромосом достигает 1% всех хромосомных нарушений у человека. Эти нарушения имеют большое эволюционное значение, ибо создают популяционное разнообразие кариотипов, генотипов и фенотипов человека. Причем каждый патологический вариант является уникальным продуктом эволюции.
В результате второго мейотического деления образуются 4 дочерние клетки. В каждую из них отойдет по одной либо материнской, либо отцовской хромосоме из всех 23 хромосом.
Чтобы избежать возможных ошибок в наших дальнейших расчетах, примем за правило: в результате второго мейотического деления также образуется 8 млн вариантов мужских гамет и 8 млн вариантов женских гамет. Тогда ответ на вопрос, каков общий объем вариантов комбинаций хромосом и расположенных на них генов при встрече двух гамет, следующий: 2 46 или 64 χ 10 12 , т.е. 64 триллиона.
Образование такого (теоретически возможного) количества генотипов при встрече двух гамет наглядно объясняет смысл гетерогенности генотипов.
Значение комбинативной изменчивости
Комбинативная изменчивость важна не только для гетерогенности и уникальности наследственного материала, но и для восстановления (репарации) стабильности молекулы ДНК при повреждении ее обеих нитей. Примером служит образование одноцепочечной бреши ДНК напротив нерепарированного повреждения. Появившаяся брешь не может быть безошибочно исправлена без привлечения к репарации нормальной нити ДНК.
Мутационная изменчивость
Наряду с уникальностью и гетерогенностью генотипов и фенотипов в результате комбинативной изменчивости огромный вклад в вариабельность генома и фенома человека вносит наследственная мутационная изменчивость и обусловленная ею генетическая гетерогенность.
Вариации нуклеотидных последовательностей ДНК чисто условно можно разделить на мутации и генетический полиморфизм (см. главу 2). Вместе с тем, если гетерогенность генотипов - это постоянные (нормальные) характеристики вариабельности генома, то мутационная изменчивость - это, как правило, его патология.
В пользу патологической вариабельности генома свидетельствуют, например, неравный кроссинговер, неправильное расхождение хромосом к полюсам деления при образовании дочерних клеток, наличие генетических компаундов и аллельных серий. Иными словами, наследственная комбинативная и мутационная изменчивость проявляется у человека значительным генотипическим и фенотипическим разнообразием.
Уточним терминологию и рассмотрим общие вопросы теории мутаций.
ОБЩИЕ ВОПРОСЫ ТЕОРИИ МУТАЦИЙ
Мутация есть изменение структурной организации, количества и/или функционирования наследственного материала и синтезируемых им белков. Это понятие впервые предложил Гуго де Фриз
в 1901-1903 гг. в своей работе «Мутационная теория», где описал основные свойства мутаций. Они:
Возникают внезапно;
Передаются из поколения в поколение;
Наследуются по доминантному типу (проявляются у гетерозигот и гомозигот) и рецессивному типу (проявляются у гомозигот);
Не имеют направленности («мутирует» любой локус, вызывая незначительные изменения или затрагивая жизненно важные признаки);
По фенотипическому проявлению бывают вредными (большинство мутаций), полезными (крайне редко) или безразличными;
Возникают в соматических и половых клетках.
Кроме того, одни и те же мутации могут возникнуть повторно.
Мутационный процесс или мутагенез, есть непрерывно идущий процесс формирования мутаций под действием мутагенов - факторов среды, повреждающих наследственный материал.
Впервые теория непрерывно идущего мутагенеза предложена в 1889 г. русским ученым из Петербургского университета С.И. Коржинским в его книге «Гетерогенезис и эволюция».
Как принято считать в настоящее время, мутации способны проявиться спонтанно, без видимых внешних причин, но под влиянием внутренних условий в клетке и организме - это спонтанные мутации или спонтанный мутагенез.
Мутации, вызванные искусственно путем воздействия внешних факторов физической, химической или биологической природы, - это индуцированные мутации, или индуцированный мутагенез.
Наиболее часто встречающиеся мутации называются мажорными мутациями (например, мутации в генах миодистрофии Дюшенна- Беккера, муковисцидоза, серповидноклеточной анемии, фенилкетонурии и др.). Сейчас созданы коммерческие наборы, позволяющие выявлять в автоматическом режиме наиболее важные из них.
Вновь возникшие мутации называются новыми мутациями или мутациями de novo. Например, к ним относятся мутации, лежащие в основе ряда аутосомно-доминантных болезней, таких, как ахондроплазия (10% случаев заболевания - семейные формы), нейрофиброматоз Реклингаузена I типа (50-70% - семейные формы), болезнь Альцгеймера, хорея Гентингтона.
Мутации от нормального состояния гена (признака) к патологическому состоянию называются прямыми.
Мутации от патологического состояния гена (признака) к нормальному состоянию называются обратными или реверсиями.
Впервые способность к реверсии установлена в 1935 г. Н.В. Тимофеевым-Рессовским.
Последующие мутации в гене, подавляющие первичный мутантный фенотип, называются супрессорными. Супрессия может быть внутригенной (восстанавливает функциональную активность белка; аминокислота не соответствует исходной, т.е. истинной обратимости нет) и внегенной (изменяется структура тРНК, в результате чего мутантная тРНК включает в полипептид другую аминокислоту вместо кодируемой дефектным триплетом).
Мутации в соматических клетках называются соматическими мутациями. Они формируют патологические клеточные клоны (совокупность патологических клеток) и в случае одновременного присутствия в организме нормальных и патологических клеток приводят к клеточному мозаицизму (например, при наследственной остеодистрофии Олбрайта экспрессивность заболевания зависит от количества аномальных клеток).
Соматические мутации могут быть как семейными, так и спорадическими (несемейными). Они лежат в основе развития злокачественных новообразований и процессов преждевременного старения.
Ранее считалось аксиомой, что соматические мутации не наследуются. В последние же годы была доказана передача из поколения в поколение наследственной предрасположенности 90% мультифакториальных форм и 10% моногенных форм рака, проявляющихся мутациями в соматических клетках.
Мутации в половых клетках называются герминативными мутациями. Считается, что они встречаются реже соматических мутаций, лежат в основе всех наследственных и некоторых врожденных болезней, передаются из поколения в поколение и также могут быть семейными и спорадическими. Наиболее изученная область общего мутагенеза - физический и, в частности, радиационный мутагенез. Любые источники ионизирующей радиации пагубны для здоровья человека, они, как правило, оказывают мощное мутагенное, тератогенное и канцерогенное воздействие. Мутагенный эффект однократной дозы облучения гораздо выше, чем при хроническом облучении; доза облучения в 10 рад удваивает частоту мутаций у человека. Доказано: ионизирующее излучение способно вызвать мутации, приводящие
к наследственным (врожденным) и онкологическим болезням, а ультрафиолетовое - индуцировать ошибки репликации ДНК.
Большую опасность представляет химический мутагенез. В мире существует около 7 млн химических соединений. В народном хозяйстве, на производстве и в быту постоянно применяются примерно 50-60 тыс. химических веществ. Ежегодно внедряются в практику около одной тысячи новых соединений. Из них 10% в состоянии индуцировать мутации. Таковы гербициды и пестициды (доля мутагенов среди них достигает 50%), а также ряд лекарственных препаратов (некоторые антибиотики, синтетические гормоны, цитостатики и др.).
Существует еще биологический мутагенез. К биологическим мутагенам относятся: чужеродные белки вакцин и сывороток, вирусы (ветряная оспа, коревая краснуха, полиомиелит, простой герпес, СПИД, энцефалит) и ДНК, экзогенные факторы (неполноценное белковое питание), соединения гистамина и его производные, стероидные гормоны (эндогенные факторы). Усиливают действие внешних мутагенов комутагены (токсины).
В истории генетики немало примеров значения связей между генами и признаками. Один из них - классификация мутаций в зависимости от их фенотипического эффекта.
Классификация мутаций в зависимости от их фенотипического эффекта
Такую классификацию мутаций впервые предложил в 1932 г. Г. Мёллер. Согласно классификации были выделены:
Аморфные мутации. Это состояние, при котором признак, контролируемый патологическим аллелем, не проявляется, так как патологический аллель не активен по сравнению с нормальным аллелем. К таким мутациям относятся ген альбинизма (11q14.1) и около 3000 аутосомно-рецессивных заболеваний;
Антиморфные мутации. В этом случае значение признака, контролируемого патологическим аллелем, противоположно значению признака, контролируемого нормальным аллелем. К таким мутациям относятся гены около 5-6 тыс. аутосомно-доминантных заболеваний;
Гиперморфные мутации. В случае такой мутации признак, контролируемый патологическим аллелем, выражен сильнее признака, контролируемого нормальным аллелем. Пример - гете-
розиготные носители генов болезней нестабильности генома (см. главу 10). Их число составляет около 3% населения Земли (почти 195 млн человек), а количество самих заболеваний достигает 100 нозологий. Среди этих заболеваний: анемия Фанкони, атаксиятелеангиэктазия, пигментная ксеродерма, синдром Блума, прогероидные синдромы, многие формы рака и др. При этом частота рака у гетерозиготных носителей генов этих заболеваний в 3-5 раз выше, чем в норме, а у самих больных (гомозигот по этим генам) частота рака в десятки раз выше, чем в норме.
Гипоморфные мутации. Это состояние, при котором проявление признака, контролируемого патологическим аллелем, ослаблено по сравнению с признаком, контролируемым нормальным аллелем. К таким мутациям относятся мутации генов синтеза пигментов (1q31; 6p21.2; 7p15-q13; 8q12.1; 17p13.3; 17q25; 19q13; Xp21.2; Xp21.3; Xp22), а также более 3000 форм аутосомно-рецессивных заболеваний.
Неоморфные мутации. О такой мутации говорят, когда признак, контролируемый патологическим аллелем, будет иного (нового) качества по сравнению с признаком, контролируемым нормальным аллелем. Пример: синтез новых иммуноглобулинов в ответ на проникновение в организм чужеродных антигенов.
Говоря о непреходящем значении классификации Г. Мёллера, следует отметить, что спустя 60 лет после ее публикации фенотипические эффекты точковых мутаций были разделены на разные классы в зависимости от оказываемого ими воздействия на структуру белкового продукта гена и/или уровень его экспрессии.
В частности, нобелевский лауреат Виктор Маккьюсик (1992) выделил мутации, изменяющие последовательность аминокислот в белке. Оказалось, что именно они отвечают за проявление 50-60% случаев моногенных болезней, а остальные мутации (40-50% случаев) приходятся на долю мутаций, затрагивающих экспрессию генов.
Изменение аминокислотного состава белка проявляется в патологическом фенотипе, например, в случаях метгемоглобинемии или серповидноклеточной анемии, обусловленной мутациями бетаглобинового гена. В свою очередь, были выделены мутации, затрагивающие нормальную экспрессию гена. Они приводят к изменению количества генного продукта и проявляются фенотипами, связанными с недостаточностью того или иного белка, например,
в случаях гемолитической анемии, обусловленной мутациями генов, локализованных на аутосомах: 9q34.3 (дефицит аденилаткиназы); 12p13.1 (дефицит триозофосфатизомеразы); 21q22.2 (дефицит фосфофруктокиназы).
Классификация мутаций В. Маккьюсика (1992) - это, безусловно, новое поколение классификаций. Вместе с тем, накануне ее публикации широкое признание получила классификация мутаций в зависимости от уровня организации наследственного материала.
Классификация мутаций в зависимости от уровня организации наследственного материала
Классификация включает следующее.
Точковые мутации (нарушение структуры гена в разных его точках).
Строго говоря, к точковым мутациям относятся изменения нуклеотидов (оснований) одного гена, ведущие к изменению количества и качества синтезируемых ими белковых продуктов. Изменения оснований - это их замены, вставки, перемещения или выпадения, которые можно объяснить мутациями в регуляторных областях генов (промотор, сайт полиаденилирования), а также в кодирующих и некодирующих областях генов (экзоны и интроны, сайты сплайсинга). Замены оснований ведут к появлению трех типов мутантных кодонов: миссенс-мутации, нейтральные мутации и нонсенс-мутации.
Точковые мутации наследуются как простые менделевские признаки. Они часто встречаются: 1 случай на 200-2000 рождений - это первичный гемохроматоз, неполипозный рак толстой кишки, синдром Мартина-Белл и муковисцидоз.
Точковые мутации, встречающиеся крайне редко (1:1 500 000), - это тяжелый комбинированный иммунодефицит (ТКИД) в результате дефицита аденозиндезаминазы. Иногда точковые мутации формируются не при воздействии мутагенов, а как ошибки репликации ДНК. При этом их частота не превышает 1:10 5 -1:10 10 , так как они исправляются с помощью репарационных систем клетки почти на
Структурные мутации или аберрации хромосом (нарушают структуру хромосом и приводят к формированию новых групп сцепления генов). Это делеции (утраты), дупликации (удвоения), транслокации (перемещения), инверсии (поворот на 180°) или инсерции (вставки) наследственного материала. Такие мутации характерны для сомати-
ческих клеток (включая стволовые клетки). Их частота составляет 1 на 1700 клеточных делений.
Известен ряд синдромов, обусловленных структурными мутациями. Наиболее известные примеры: синдром «кошачьего крика» (кариотип: 46,ХХ,5р-), синдром Вольфа-Хиршхорна (46,ХХ, 4р-), транслокационная форма синдрома Дауна (кариотип: 47, ХУ, t (14;21)).
Другой пример - это лейкемии. При них происходит нарушение экспрессии гена в результате так называемого разделения (транслокация между структурной частью гена и его промоторной областью), и, следовательно, нарушается синтез белка.
Геномные (численные) мутации - нарушение числа хромосом или их частей (ведут к появлению новых геномов или их частей путем добавления или утраты целых хромосом или их частей). Происхождение этих мутаций обусловлено нерасхождением хромосом в митозе или мейозе.
В первом случае - это анеуплоиды, тетраплоиды с неразделенной цитоплазмой, полиплоиды, имеющие по 6, 8, 10 пар хромосом и более.
Во втором случае - это неразделение парных хромосом, участвующих в формировании гамет (моносомии, трисомии) или оплодотворение одной яйцеклетки двумя сперматозоидами (диспермия или триплоидный зародыш).
Их типичные примеры уже не раз приводились - это синдром Шерешевского-Тернера (45,ХО), синдром Клайнфельтера (47,ХХУ), регулярная трисомия при синдроме Дауна (47,ХХ, +21).



