Приращение энтропии. Энтропия
Второй закон термодинамики в виде , записанный для равновесных процессов, позволяет вычислить не абсолютное значение энтропии, а только разность энтропий в двух состояниях системы.
 . (2.4)
. (2.4)
Рассмотрим для 1 моля вещества :
а) Изотермические процессы (T = const ).
При
постоянной температуре протекают
процессы фазовых превращений веществ:
плавление, испарение и другие. При
равновесном протекании этих процессов
давление сохраняется обычно постоянным,
поэтому
 и
и
 , (2.5)
, (2.5)
где
 – энтальпия фазового перехода.
– энтальпия фазового перехода.
б) Изобарные процессы (р = const ).
Если нагревание происходит при постоянном давлении, то
 , (2.6)
, (2.6)
где n – число молей вещества. Тогда
 . (2.7)
. (2.7)
Пример 2.1. Определить изменение энтропии при нагреве 1 моль Al от 25 до 600 0 С, если для него в этом интервале теплоёмкость зависит от температуры следующим образом:
 ,
(Дж/моль К).
,
(Дж/моль К).
Решение. Согласно уравнению (2.7) имеем:
 ,
,
(Дж/моль К).
с) Изохорные процессы (V = const ).
Если нагревание происходит при постоянном объёме, то
 . (2.8)
. (2.8)
 . (2.9)
. (2.9)
Для 1 моля идеального газа справедливо :
а) При изменении объёма и температуры
 , (2.10)
, (2.10)
с
учетом, что
 .
.
б) При изменении давления и температуры
 . (2.11)
. (2.11)
Для любого вещества при любой температуре можно определить и абсолютное значение энтропии, если воспользоваться постулатом Планка : энтропия правильно образованного кристалла любого индивидуального вещества при абсолютном нуле равна нулю.
Если вещество при температуре Т находится в газообразном состоянии, то его абсолютная энтропия может быть вычислена по формуле:
2.2.2. Расчёт изменения энтропии в ходе химической реакции.
Расчёт изменения энтропии в ходе химической реакции проводится по формуле:
где
 - стандартные энтропии веществ приТ
= 298,15 К.
- стандартные энтропии веществ приТ
= 298,15 К.
Каждое
вещество характеризуется стандартной
энтропией
 – энтропией 1 моль вещества при 298.15 К
и давлении 1 атм. Значения энтропии
имеют размерность Дж/(моль К) или кал/(моль
К).Стандартные
энтропии простых веществ не равны нулю.
– энтропией 1 моль вещества при 298.15 К
и давлении 1 атм. Значения энтропии
имеют размерность Дж/(моль К) или кал/(моль
К).Стандартные
энтропии простых веществ не равны нулю.
2.2.3. Расчёт изменения энтропии в ходе самопроизвольных (необратимых) процессов.
Для
необратимых процессов
 и уравнение (2.4) не применимо. Энтропия
– функция состояния и её изменение не
зависит от пути процесса, а определяется
конечным и начальным состоянием системы.
Изменение энтропии в любом неравновесном
процессе можно вычислить, заменяя его
некоторой совокупностью равновесных
процессов, просходящими между теми же
начальными и конечными состояниями,
для каждого из которых можно рассчитать
значение
и уравнение (2.4) не применимо. Энтропия
– функция состояния и её изменение не
зависит от пути процесса, а определяется
конечным и начальным состоянием системы.
Изменение энтропии в любом неравновесном
процессе можно вычислить, заменяя его
некоторой совокупностью равновесных
процессов, просходящими между теми же
начальными и конечными состояниями,
для каждого из которых можно рассчитать
значение .
Тогда:
.
Тогда:
 . (2.14)
. (2.14)
2.3. Энергия гиббса, энергия гельмгольца. Уравнение гиббса–гельмгольца.
В изолированных системах энтропия только увеличивается и при равновесии достигает максимума. Поэтому она может быть использована в качестве критерия протекания самопроизвольных процессов в таких системах. Однако на практике большинство процессов происходит в неизолированных системах, вследствие чего для них надо выбрать свои критерии направления самопроизвольных процессов и достижения равновесия в этих системах. Такими критериями являются другие термодинамические функции, отличные от энтропии и внутренней энергии. Они подобраны таким образом, что с их помощью можно определить в явной форме все термодинамические параметры изучаемой системы. Все они являются функциями состояния и при переходе системы из одного положения в другое меняются однозначно. При достижении системой равновесного состояния каждая из функций проходит через минимальное значение. Такие свойства обуславливают широкое применение этих функций при аналитическом методе решения различных задач термодинамических исследований.
Следует отметить, что такие функции часто называют характеристическими. Характеристической функцией называется такая функция состояния системы, посредством которой и её производных могут быть выражены в явной форме все термодинамические свойства системы.
Согласно первому закону термодинамики:
A = Q – dU . (2.15)
Подставив сюда извесное соотношение Q ≤ TdS, получим
A ≤ TdS – dU , (2.16)
где знак равенства относится к обратимым равновесным процессам, а знак неравенства - к необратимым. Проинтегрируем (2.16) при Т = const :
A T ≤ T (S 2 – S 1) – (U 2 – U 1) = (U 1 – TS 1) – (U 2 – TS 2). (2.17)
Функция (U – TS ) играет большую роль при изучении равновесия в изотермических процессах. Её называют изохорно-изотермическим потенциалом или энергией Гельмгольца и обозначают символом F . При этом для всякого изотермического процесса:
dF = dU – TdS , (2.18)
∆F = ∆U – T∆S , (2.19)
а максимальная работа в изотермическом процессе
(A Т ) max = –∆F . (2.20)
Функция F определяет направление и предел течения самопроизвольных процессов, протекающих при постоянных температуре и объёме.
Близкой к изохорно-изотермическому потенциалу является функция, определяющая направление и предел самопроизвольного протекания процессов для систем, находящихся при постоянных температуре и давлении. Эта функция называется изобарно-изотер-мическим потенциалом или энергией Гиббса , обозначается символом G и определяется как
G = H – TS . (2.21)
G = U – TS + pV = F + pV . (2.22)
Пусть р = const, тогда
A T ≤ –∆F = F 1 – F 2 , (2.23)
A T + p (V 2 – V 1) ≤ F 1 – F 2 , (2.24)
A T ≤ (F 1 +pV 1) – (F 2 + pV 2) = G 1 – G 2 , (2.25)
где A T – полезная работа (любая работа кроме работы расширения). Тогда
A T ≤ –∆G . (2.26)
При этом для изотермических процессов
 , (2.27)
, (2.27)
и максимальная работа в изотермическом процессе
 , (2.29)
, (2.29)
т.е. максимальная полезная работа равна максимальной работе изотермического процесса за вычетом работы против сил внешнего давления. Функции G и F называются термодинамическими потенциалами , потому что в определённых условиях стремятся к минимуму при протекании самопроизвольных процессов.
Пусть
 ,
тогда
,
тогда
 . (2.30)
. (2.30)
1). Система
при T
, V
= const
 ,
т. е. ΔF
≤ 0.
Условие равновесия в изохорно-изотермической
системе: dF
= 0,
ΔF
= 0,
F
= F
min .
,
т. е. ΔF
≤ 0.
Условие равновесия в изохорно-изотермической
системе: dF
= 0,
ΔF
= 0,
F
= F
min .
2). Система
при р
, T
= const
.
Тогда
 Условие равновесия в изобарно-изотерми-ческой
системе:dG
= 0,
ΔG
= 0,
G
= G
min .
Условие равновесия в изобарно-изотерми-ческой
системе:dG
= 0,
ΔG
= 0,
G
= G
min .
Вывод: в системах, находящихся при постоянных температуре и объёме, самопроизвольно могут протекать только те процессы, которые сопровождаются уменьшением энергии Гельмгольца F , причем пределом их протекания, т.е. условием равновесия, является достижение некоторого минимального для данных условий значения функции F ; в системах же, находящихся при постоянных температуре и давлении, самопроизвольно могут протекать только те процессы, которые сопровождаются уменьшением энергии Гиббса G , причём пределом их протекания, т.е. условием равновесия, служит достижение некоторого минимального для данных условий значения функции G .
Получим соотношения,
которые описывают зависимость
 и
и от температуры. В общем случае (и для
химических реакций):
от температуры. В общем случае (и для
химических реакций):
Функция
состояния обладает свойствами полного
дифференциала, т.е. если
 ,
то
,
то
 . (2.33)
. (2.33)
С другой стороны:
F = U – TS , (2.34)
dF = dU – TdS – SdT . (2.35)
С учетом того, что
dU = , (2.36)
получаем
 . (2.37)
. (2.37)
При сравнении уравнений (2.37) и (2.33) видно, что

 , (2.38)
, (2.38)
 . (2.39)
. (2.39)
Аналогично для
 ,
получаем:
,
получаем:
 , (2.40)
, (2.40)

 , (2.41)
, (2.41)
 . (2.42)
. (2.42)
Подставляя соотношения (2.39) и (2.42) в уравнения (2.31) и (2.32), соответственно, получаем:
 , (2.43)
, (2.43)
 . (2.44)
. (2.44)
Последние
два равенства и есть искомые зависимости
 и
и от температуры и их называютуравнениями
Гиббса-Гельмгольца
.
от температуры и их называютуравнениями
Гиббса-Гельмгольца
.
Второй закон термодинамики устанавливает критерии необратимости термодинамических процессов. Известно много формулировок второго закона, которые эквивалентны друг другу. Мы приведем здесь только одну формулировку, связанную с энтропией.
Существует функция состояния - энтропия S , которая обладает следующим свойством: , (4.1) где знак равенства относится к обратимым процессам, а знак больше - к необратимым.
Для изолированных систем второй закон
утверждает: dS
і 0, (4.2) т.е. энтропия
изолированных систем в необратимых процессах
может только возрастать, а в состоянии
термодинамического равновесия она достигает
максимума (dS
= 0,
d
2 S
< 0).
Неравенство (4.1) называют неравенством Клаузиуса . Поскольку энтропия - функция состояния, ее изменение в любом циклическом процессе равно 0, поэтому для циклических процессов неравенство Клаузиуса имеет вид:
где знак равенства ставится, если весь цикл полностью обратим.
Энтропию можно определить с помощью двух эквивалентных подходов - статистического и термодинамического. Статистическое определение основано на идее о том, что необратимые процессы в термодинамике вызваны переходом в более вероятное состояние, поэтому энтропию можно связать с вероятностью:
где k = 1.38 10 -23 Дж/К - постоянная Больцмана (k = R / N A), W - так называемая термодинамическая вероятность, т.е. число микросостояний, которые соответствуют данному макросостоянию системы (см. гл. 10). Формулу (4.4) называют формулой Больцмана .
С точки зрения строгой статистической термодинамики энтропию вводят следующим образом:
где G (E ) - фазовый объем, занятый микроканоническим ансамблем с энергией E .
Термодинамическое определение энтропии основано на рассмотрении обратимых процессов:
Это определение позволяет представить элементарную теплоту в такой же форме, как и различные виды работы:
Q обр = TdS , (4.7)
где температура играет роль обобщенной силы, а энтропия - обобщенной (тепловой) координаты.
Расчет изменения энтропии для различных процессов
Термодинамические расчеты изменения энтропии основаны на определении (4.6) и на свойствах частных производных энтропии по термодинамическим параметрам:
 (4.8)
(4.8)
Последние два тождества представляют собой соотношения Максвелла (вывод см. в гл. 5).
1) Нагревание или охлаждение при постоянном давлении .
Количество теплоты, необходимое для изменения температуры системы, выражают с помощью теплоемкости: Q обр = C p dT .
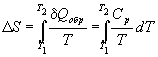 (4.9)
(4.9)
Если теплоемкость не зависит от температуры в интервале от T 1 до T 2 , то уравнение (4.8) можно проинтегрировать:
Если изменение температуры происходит при постоянном объеме, то в формулах (4.9) и (4.10) C p надо заменить на C V .
2) Изотермическое расширение или сжатие .
Для расчета энтропии в этом случае надо знать уравнение состояния системы. Расчет основан на использовании соотношения Максвелла:
 (4.11)
(4.11)
В частности, для изотермического расширения идеального газа (p = nRT / V )
Этот же результат можно получить, если использовать выражение для теплоты изотермического обратимого расширения идеального газа: Q обр = nRT ln(V 2 /V 1) .
3) Фазовые переходы .
При обратимом фазовом переходе температура остается постоянной, а теплота фазового перехода при постоянном давлении равна H фп, поэтому изменение энтропии равно:
![]() (4.13)
(4.13)
При плавлении и кипении теплота поглощается, поэтому энтропия в этих процессах возрастает: S тв < S ж < S г. При этом энтропия окружающей среды уменьшается на величину S ф.п. , поэтому изменение энтропии Вселенной равно 0, как и полагается для обратимого процесса в изолированной системе.
4) Смешение идеальных газов при постоянных температуре и давлении .
Если n 1 молей одного газа, занимающего объем V 1 , смешиваются с n 2 молями другого газа, занимающего объем V 2 , то общий объем будет равен V 1 + V 2 , причем газы расширяются независимо друг от друга и общее изменение энтропии равно сумме изменений энтропии каждого газа:
где x i - мольная доля i -го газа в полученной газовой смеси. Изменение энтропии (4.14) всегда положительно, т.к. все ln x i < 0, поэтому идеальные газы всегда смешиваются необратимо.
Если при тех же условиях смешиваются две порции одного и того же газа, то уравнение (4.14) уже неприменимо. Никаких изменений в системе при смешивании не происходит, и S = 0. Тем не менее, формула (4.14) не содержит никаких индивидуальных параметров газов, поэтому, казалось бы, должна быть применима и к смешению одинаковых газов. Это противоречие называют парадоксом Гиббса .
Абсолютная энтропия
В отличие от многих других термодинамических функций, энтропия имеет точку отсчета, которая задается постулатом Планка (третьим законом термодинамики) :
При абсолютном нуле T = 0 К все идеальные кристаллы
имеют одинаковую энтропию, равную нулю.
При стремлении температуры к абсолютному нулю не только энтропия стремится к 0, но и ее производные по всем термодинамическим параметрам:
(x = p , V ). (4.15)
Это означает, что вблизи абсолютного нуля все термодинамические процессы протекают без изменения энтропии. Это утверждение называют тепловой теоремой Нернста .
Постулат Планка позволяет ввести понятие абсолютной энтропии вещества, т.е. энтропии, отсчитанной от нулевого значения при T = 0. Для расчета абсолютной энтропии веществ в стандартном состоянии надо знать зависимости теплоемкости C p от температуры для каждой из фаз, а также температуры и энтальпии фазовых переходов. Так, например, абсолютная энтропия газообразного вещества в стандартном состоянии при температуре T складывается из следующих составляющих:
В термодинамических таблицах обычно приводят значения абсолютной энтропии в стандартном состоянии при температуре 298 К.
Значения абсолютной энтропии веществ используют для расчета изменения энтропии в химических реакциях:
![]() . (4.17)
. (4.17)
ПРИМЕРЫ
Пример 4-1. Определите зависимость энтропии от объема для термодинамической системы, которая описывается уравнением состояния (для одного моля)
![]()
Решение .
![]()
Интегрируя это равенство, находим зависимость энтропии от объема:
где const зависит от температуры.
Пример 4-2. Рассчитайте изменение энтропии при нагревании 0.7 моль моноклинной серы от 25 до 200 о С при давлении 1 атм. Мольная теплоемкость серы равна:
C p (S тв) = 23.64 Дж/(моль.
К),
C p
(S ж) = 35.73 + 1.17 . 10 -3 . T
Дж/(моль. К).
Температура плавления моноклинной серы 119 о С, удельная теплота плавления 45.2 Дж/г.
Решение . Общее изменение энтропии складывается из трех составляющих: 1) нагревание твердой серы от 25 до 119 о С, 2) плавление, 3) нагревание жидкой серы от 119 до 200 о С.
 4.54 Дж/К.
4.54 Дж/К.
![]() 2.58 Дж/К.
2.58 Дж/К.

S = S 1 + S 2 + S 3 = 11.88 Дж/К.
Ответ. 11.88 Дж/К.
Пример 4-3. Найдите изменение энтропии газа и окружающей среды, если n молей идеального газа расширяются изотермически от объема V 1 до объема V p .
Решение . а) Изменение энтропии газа при обратимом изотермическом расширении можно найти с помощью термодинамического определения энтропии с расчетом теплоты расширения по первому закону:
 .
.
Так как расширение обратимое, то общее изменение энтропии Вселенной равно 0, поэтому изменение энтропии окружающей среды равно изменению энтропии газа с обратным знаком:
![]() .
.
б) Энтропия - функция состояния, поэтому изменение энтропии системы не зависит от того, как совершался процесс - обратимо или необратимо. Изменение энтропии газа при необратимом расширении против внешнего давления будет таким же, как и при обратимом расширении. Другое дело - энтропия окружающей среды, которую можно найти, рассчитав с помощью первого закона теплоту, переданную системе:
![]() .
.
В этом выводе мы использовали тот факт, что U = 0 (температура постоянна). Работа, совершаемая системой против постоянного внешнего давления равна: A = p (V 2 -V 1), а теплота, принятая окружающей средой, равна работе, совершенной системой, с обратным знаком.
Общее изменение энтропии газа и окружающей среды больше 0:
![]() ,
,
как и полагается для необратимого процесса.
Пример 4-4. Рассчитайте изменение энтропии 1000 г воды в результате ее замерзания при -5 О С. Теплота плавления льда при 0 о С равна 6008 Дж/моль. Теплоемкости льда и воды равны 34.7 и 75.3 Дж/(моль. К), соответственно. Объясните, почему энтропия при замерзании уменьшается, хотя процесс - самопроизвольный.
Решение
. Необратимый процесс замерзания
воды при температуре -5 О С можно
представить в виде последовательности обратимых
процессов: 1) нагревание воды от
-5 О С до температуры замерзания (0 О С);
2) замерзание воды при 0 О С; 3) охлаждение
льда от 0 до -5 О С:

Изменение энтропии в первом и третьем процессах (при изменении температуры) рассчитывается по формуле (4.9):
 77.3 Дж/К.
77.3 Дж/К.
 -35.6 Дж/К.
-35.6 Дж/К.
Изменение энтропии во втором процессе рассчитывается как для обычного фазового перехода (4.13). Необходимо только иметь в виду, что теплота при замерзании выделяется:
![]() -1223 Дж/К.
-1223 Дж/К.
Т.к. энтропия - функция состояния, общее изменение энтропии равно сумме по этим трем процессам:
S = S 1 + S 2 + S 3 = -1181 Дж/К.
Энтропия при замерзании убывает, хотя процесс самопроизвольный. Это связано с тем, что в окружающую среду выделяется теплота и энтропия окружающей среды увеличивается, причем это увеличение больше, чем 1181 Дж/К, поэтому энтропия Вселенной при замерзании воды возрастает, как и полагается в необратимом процессе.
Ответ. -1181 Дж/К.
ЗАДАЧИ
4-1. Приведите пример термодинамического процесса, который может быть проведен как обратимо, так и необратимо. Рассчитайте изменение энтропии системы и окружающей среды в обоих случаях.
4-2. Проверьте неравенство Клаузиуса для циклического процесса, представленного в задаче 2.14.
4-3. Рассчитайте мольную энтропию неона при 500 К, если при 298 К и том же объеме энтропия неона равна 146.2 Дж/(моль. К).
4-4. Рассчитайте изменение энтропии при нагревании 11.2 л азота от 0 до 50 о С и одновременном уменьшении давления от 1 атм до 0.01 атм.
4-5. Один моль гелия при 100 о С и 1 атм смешивают с 0.5 моль неона при 0 о С и 1 атм. Определите изменение энтропии, если конечное давление равно 1 атм.
4-6. Рассчитайте изменение энтропии при образовании 1 м 3 воздуха из азота и кислорода (20 об.%) при температуре 25 о С и давлении 1 атм.
4-7. Три моля идеального одноатомного газа (C V = 3.0 кал/(моль. К)), находящегося при T 1 = 350 K и P 1 = 5.0 атм, обратимо и адиабатически расширяются до давления P 2 = 1.0 атм. Рассчитайте конечные температуру и объем, а также совершенную работу и изменение внутренней энергии, энтальпии и энтропии в этом процессе.
4-8. Рассчитайте изменение энтропии при нагревании 0.4 моль хлорида натрия от 20 до 850 о С. Мольная теплоемкость хлорида натрия равна:
C p (NaCl тв) = 45.94 + 16.32 . 10 -3 .
T
Дж/(моль. К),
C p
(NaCl ж) = 66.53 Дж/(моль. К).
Температура плавления хлорида натрия 800 о С, теплота плавления 31.0 кДж/моль.
4-9. Рассчитайте изменение энтропии при смешении 5 кг воды при 80 о С с 10 кг воды при 20 о С. Удельную теплоемкость воды принять равной: C p (H 2 O) = 4.184 Дж/(г. К).
4-10. Рассчитайте изменение энтропии при добавлении 200 г льда, находящегося при температуре 0 о С, к 200 г воды (90 о С) в изолированном сосуде. Теплота плавления льда равна 6.0 кДж/моль.
4-11. Для некоторого твердого тела найдена зависимость коэффициента расширения от давления в интервале давлений от p 1 до p 2:
![]() .
.
Насколько уменьшится энтропия этого тела при сжатии от p 1 до p 2 ?
4-12. Найдите изменение энтропии газа и окружающей среды, если n молей идеального газа расширяются изотермически от давления p 1 до давления p 2: а) обратимо; б) против внешнего давления p < p 2 .
4-13. Запишите выражение для расчета абсолютной энтропии одного моля воды при температуре 300 0 С и давлении 2 атм.
4-14. Нарисуйте график зависимости стандартной энтропии воды от температуры в интервале от 0 до 400 К.
4-15. Запишите энтропию одного моля идеального газа как функцию температуры и давления (теплоемкость считать постоянной).
4-16. Определите зависимость энтропии от объема для термодинамической системы, которая описывается уравнением состояния (для одного моля):
![]()
4-17. Определите зависимость энтропии от объема для термодинамической системы, которая описывается уравнением состояния (для одного моля):
![]()
4-18. Один моль газа описывается уравнением состояния
где f (V ) - некоторая функция, которая не зависит от температуры. Рассчитайте изменение энтропии газа при его необратимом изотермическом расширении от объема V 1 до объема V 2 .
4-19. Рассчитайте изменение энтропии 1000 г метанола в результате его замерзания при -105 О С. Теплота плавления твердого метанола при -98 о С (т.пл.) равна 3160 Дж/моль. Теплоемкости твердого и жидкого метанола равны 55.6 и 81.6 Дж/(моль. К), соответственно. Объясните, почему энтропия при замерзании уменьшается, хотя процесс - самопроизвольный.
4-20. Теплоемкость некоторого вещества в интервале температур от T 1 до T 2 изменяется следующим образом:

Постройте график зависимости энтропии вещества от температуры в этом интервале температур.
4-21. Пользуясь справочными данными, приведите пример самопроизвольной химической реакции, для которой стандартное изменение энтропии меньше 0.
4-22. Пользуясь справочными данными, рассчитайте стандартное изменение энтропии в реакции H 2(г) + ЅO 2(г) = H 2 O (г) а) при 25 о С; б) при 300 о С.
Математическое выражение второго закона термодинамики записывается:
Здесь знак > относится к необратимым процессам, а знак = к обратимым. Так как энтропия является функцией состояния, ее изменение при протекании как обратимого, так и необратимого процессов одинаково. Поэтому при расчете изменения энтропии необходимо пользоваться формулами для обратимых процессов.
Энтропия обладает свойствами аддитивности, поэтому изменение энтропии в сложном процессе равно сумме изменений энтропий в отдельных его стадиях. Абсолютное значение энтропии какого-либо вещества при любой температуре можно рассчитать, если известна абсолютная энтропия при какой-то одной температуре, например, при 298К и температурные коэффициенты теплоемкости:
Изменение энтропии в различных процессах вычисляют по следующим уравнениям:
При нагревании n – моль вещества от Т 1 до Т 2 при P = const:
Интегрирование дает:
При фазовом превращении:
Где λ – молярная теплота фазового перехода (плавления, испарения, сублимации, модификационного превращения); Т – температура фазового перехода.
При переходе n – моль идеального газа из состояния 1 в состояние 2 при Т=const:
При смешении идеальных газов (Т,Р=const):
Где n 1 и n 2 – числа моль первого и второго газа: V 1 и V 2 – их начальные объемы:
V= V 1 + V 2 - конечный объем.
Определить изменение энтропии при превращении 2г льда, взятого при температуре 253К и давлении 1,013*10 5 н/м 2 в пар при температуре 423К, если теплота плавления льда при 273К равна 0,335 кДж/г, удельная теплоемкость льда равна 2,02 Дж/г*К воды – 4,2 Дж/г. К, скрытая теплота парообразования воды равна 2,255 кДж/г, мольная теплоемкость пара при постоянном давлении:
С р = 30,13+11,3*10 -3 Т, Дж/моль. К
Данный процесс состоит из пяти стадий:
1) нагревание льда от 253 до 273 К – ∆S 1 ;
2) плавление льда при 273 К – ∆S 2 ;
3) нагревание жидкой воды от 273 до 373 К – ∆S 3 ;
4) переход жидкой воды в пар при 373К – ∆S 4 ;
5) нагревание водяного пара от 373 до 473 К – ∆S 5 .
В одном из сосудов вместимостью 0,1 м 3 находится кислород, в другом, вместимостью 0,4 м 3 – азот. В обоих сосудах температура 290 К и давление 1,013 · 10 5 Н/м 2 . Найти изменение энтропии при смешении газов, считая их идеальными.
Находим числа моль газов по уравнению Менделеева – Клапейрона:
Вычислить стандартное изменение энтропии для реакции: Cd+2AgCl = 2Ag+CdCl 2 , если
2.2. Вычисление изменения изобарного и изохорного
потенциалов в различных процессах
В изобарно-изотермическом процессе (Р , Т = const) критерием направления процесса и равновесия является изобарно-изотермический потенциал или свободная энергия Гиббса: ∆G ≤ 0. При равновесии G минимальна. В изохорно-изотермическом процессе (V , T = const) критерием направления процесса и равновесия служит изохорно-изотермический потенциал или свободная энергия Гельмгольца: ∆F ≤ 0. При равновесии F минимальна.
Изменения ∆G и ∆F при постоянной температуре рассчитываются по формулам: ∆G = ∆H – T ∆S и ∆F = ∆U – T ∆S .
Из этих уравнений видно, что свободная энергия G или F являются частью полного запаса энергии системы Н или U за вычетом связанной энергии T S . Свободная энергия может быть извлечена из системы и превращена в работу: -∆G = A р макс и -∆F = = A V макс, где A р макс – максимальная полная работа; A V макс – максимальная полезная работа.
При расширении или сжатии идеального газа при постоянной температуре
Зависимость ∆G и ∆F от температуры выражается уравнением Гиббса – Гельмгольца. Для ∆G в интегральной форме оно записывается так:
или в пределах от 298 до Т :
здесь ∆Н = f (T ).
Для химической реакции
∆G = ∆F + ∆nRT ,
Если термодинамическая система перешла из одного состояния в другое, то полученное ею количество теплоты зависит не только от начального и конечного состояний, но и от вида процесса перехода. Иными словами, количество теплоты является функцией процесса, а не состояния. В поисках функции состояния рассмотрим величину, приращение которой равно отношению тепла, поглощенного на участке обратимого процесса, к температуре:
Эта величина называется энтропией или приведенным количеством теплоты. Понятие энтропии введено в 1865 г. Р. Клаузиусом. Несложно показать, что сумма всех приращений энтропии в цикле Карно равна нулю. Действительно, для адиабатных процессов 2-3 и 4-1 dS= О, поскольку dQ = 0 (рис. 13.3). А для оставшихся изотермических процессов 1-2 и 3^1 интегрирование (13.12) с учетом (13.5) и (13.7) дает
Этот результат справедлив независимо от выбора рабочего тела. Можно показать, что изменение энтропии равно нулю не только в цикле Карно, но и в любом другом обратимом цикле. Таким образом, энтропия является функцией состояния, а ее значения в начале и в конце кругового процесса одинаковы.
Можно показать, что энтропия системы, совершающей необратимый цикл, возрастает. В общем случае справедливо неравенство Клаузиуса: энтропия замкнутой системы возрастает (в случае необратимых процессов) либо остается постоянной (в случае обратимых процессов)
Найдем теперь с помощью (12.9) и (12.3) изменение энтропии в процессах идеального газа:
Здесь давление выражено с помощью уравнения Менделеева- Клапейрона. Интегрирование полученного выражения дает
Следует подчеркнуть, что изменение энтропии определяется только начальным и конечным состояниями идеального газа и не зависит от характера процесса перехода. Из формулы (13.17) следует, что при изотермическом процессе
а при изохорном процессе
Как уже отмечалось, из определения энтропии следует, что для адиабатного процесса dS = 0. Энтропия измеряется в джоулях на кельвин (Дж/К). Энтропия обладает свойством аддитивности: энтропия системы равна сумме энтропии тел, входящих в систему. Связано это с тем, что тепло, поглощенное системой, складывается из порций тепла, поглощенных ее частями. Свойством аддитивности обладают также внутренняя энергия, масса, объем (а, например, температура и давление таким свойством не обладают).
Энтропия - понятие не только термодинамическое, но и статистическое. Она связана с термодинамической вероятностью состояния системы. Термодинамическая вероятность W состояния системы - это число способов, которыми может быть реализовано данное состояние макроскопической системы, или число микросостояний, осуществляющих данное макросостояние. Термодинамическая вероятность состояния системы, состоящей всего из 10 молекул газа, примерно 1000, а в реальных системах молекул на много порядков больше. Поэтому более удобным для восприятия в термодинамике оказалось использовать не величину W, а ее логарифм In W. Последнему можно придать размерность (Дж/К), умножив на константу Больцмана к. Энтропия системы и термодинамическая вероятность связаны между собой формулой Больцмана : где к - постоянная Больцмана.
Таким образом, энтропия равна логарифму числа микросостояний, с помощью которых может быть реализовано данное макросостояние. Чем больше число микросостояний, реализующих данное макросостояние, тем больше энтропия. В состоянии равновесия - наиболее вероятного состояния системы - число микросостояний максимально, при этом максимальна и энтропия.
Часто воспринимают энтропию как меру беспорядка в системе. Связано это с тем, что упорядоченные системы обычно имеют гораздо меньше микросостояний, чем неупорядоченные. Так 100 молекул в двух половинах сосуда поровну можно разместить, в соответствии с теорией вероятности, огромным количеством способов. А размещению всех их в одной половине соответствует лишь два варианта. Поэтому и стремятся молекулы распределиться по объему примерно поровну, и энтропия при этом максимальна. Рассмотрим, например, распределение молекул идеального газа. В случае идеального газа наиболее вероятным состоянием, соответствующим максимуму энтропии, будет равномерное распределение молекул. При этом реализуется и максимальный «беспорядок», так как возможности конфигурирования будут максимальные. Реальные необратимые процессы в замкнутой системе ведут к увеличению ее энтропии - в этом состоит принцип возрастания энтропии.
Следует подчеркнуть, что вышеприведенные утверждения, в том числе и второе начало термодинамики, носят статистический характер и могут не выполняться для систем из малого числа частиц.
Энтропия
Изменение энтальпии системы не может служить единственным критерием самопроизвольного осуществления химической реакции, поскольку многие эндотермические процессы протекают самопроизвольно. Иллюстрацией этого служит растворение некоторых солей (например, NH 4NO 3) в воде, сопровождающееся заметным охлаждением раствора. Необходимо учитывать еще один фактор, определяющий способность самопроизвольно переходить из более упорядоченного к менее упорядоченному (более хаотичному) состоянию.
Энтропия (S ) – термодинамическая функция состояния, которая служит мерой беспорядка (неупорядоченности) системы. Возможность протекания эндотермических процессов обусловлена изменением энтропии, ибо в изолированных системах энтропия самопроизвольно протекающего процесса увеличивается ΔS > 0 (второй закон термодинамики ).
Л. Больцман определил энтропию как термодинамическую вероятность состояния (беспорядок) системы W . Поскольку число частиц в системе велико (число Авогадро N A = 6,02∙10 23), то энтропия пропорциональна натуральному логарифму термодинамической вероятности состояния системы W :
Размерность энтропии 1 моля вещества совпадает с размерностью газовой постоянной R и равна Дж∙моль –1∙K –1. Изменение энтропии *) в необратимых и обратимых процессах передается соотношениями ΔS > Q / T и ΔS = Q / T . Например, изменение энтропии плавления равно теплоте (энтальпии) плавления ΔS пл = ΔH пл/T пл Для химической реакции изменение энтропии аналогично изменению энтальпии
*) термин энтропия был введен Клаузиусом (1865 г.) через отношение Q/T (приведенное тепло).
Здесь ΔS ° соответствует энтропии стандартного состояния. Стандартные энтропии простых веществ не равны нулю. В отличие от других термодинамических функций энтропия идеально кристаллического тела при абсолютном нуле равна нулю (постулат Планка), поскольку W = 1.
Энтропия вещества или системы тел при определенной температуре является абсолютной величиной. В табл. 4.1 приведены стандартные энтропии S ° некоторых веществ.
|
||||||||||||||||||||||||||||||||||||||||||||
Таблица 4.1. Стандартные энтропии некоторых веществ. |
Из табл. 4.1 следует, что энтропия зависит от:
- Агрегатного состояния вещества. Энтропия увеличивается при переходе от твердого к жидкому и особенно к газообразному состоянию (вода, лед, пар).
- Изотопного состава (H 2O и D 2O).
- Молекулярной массы однотипных соединений (CH 4, C 2H 6, н-C 4H 10).
- Строения молекулы (н-C 4H 10, изо-C 4H 10).
- Кристаллической структуры (аллотропии) – алмаз, графит.
Наконец, рис. 4.3 иллюстрирует зависимость энтропии от температуры.
Следовательно, стремление системы к беспорядку проявляется тем больше, чем выше температура. Произведение изменения энтропии системы на температуру T ΔS количественно оценивает эту тендецию и называется энтропийным фактором .
Задачи и тесты по теме "Химическая термодинамика. Энтропия"
- Химические элементы. Знаки химических элементов - Первоначальные химические понятия и теоретические представления 8–9 класс
Уроков: 3 Заданий: 9 Тестов: 1



